
Autores:
Danieli Castro Oliveira de AndradeDoutora, Médica reumatologista do Centro Paulista de Investigação Clínica e do Instituto Fleury, São Paulo. (Ex-Residente da FMUSP - Reumatologia)
Eduardo Ferreira Borba
Professor Livre Docente da Disciplina de Reumatologia da Faculdade de Medicina da Universidade de São Paulo – FMUSP.
Última revisão: 14/04/2011
Comentários de assinantes: 0
INTRODUÇÃO E DEFINIÇÃO
A amiloidose
compreende um grupo heterogêneo de doenças que possuem em comum o
depósito extracelular de uma substância proteica dita amiloide. É
interessante que todas as formas de amiloide apresentam a
birrefringência verde peculiar à microscopia após a coloração pelo
vermelho-Congo, e essa observação no material de biópsia continua sendo o
padrão-ouro para estabelecer o diagnóstico de amiloidose.
As
doenças por amiloide são classificadas de acordo com a composição
bioquímica das proteínas precursoras séricas que formam as fibrilas de
amiloide e depósitos. Apesar destes avanços, sua história natural é
ainda pouco conhecida e o seu diagnóstico, na maioria das vezes,
realizado tardiamente, quando a doença já se encontra num estágio mais
avançado.
ETIOLOGIA E FISIOPATOLOGIA
As
doenças amiloides são classificadas de acordo com o padrão e a extensão
do depósito. Podem ser divididas em adquiridas ou hereditárias, ou
ainda sistêmicas ou localizadas de acordo com a extensão do depósito,
conforme Tabela 1.
As
formas mais frequentes de amiloidose são as adquiridas, onde
encontramos as formas AL (imunoglobulinas de cadeia leve ou primária),
as AA (reativa ou secundária) e as A-beta-2M (beta-2-microglobulina ou
associada à diálise). A amiloidose do tipo AL é rara, com incidência
estimada de 4,5 por 100.000 indivíduos. Por sua vez, amiloidose
secundária ou reativa (tipo AA) está principalmente associada a
infecções e inflamações crônicas, e corresponde a 8 a 16% dos casos de amiloidose. Doenças como tuberculose e hanseníase podem ter incidência superior a 50%.
Por
outro lado, as formas hereditárias são mais raras e variam de acordo
com a região geográfica, acometendo 1 em 100.000 indivíduos. Geralmente,
o modo de transmissão é autossômico dominante, com a exceção da febre
familiar do Mediterrâneo, que apresenta o padrão autossômico recessivo.
ACHADOS CLÍNICOS
As apresentações clínicas variam de acordo com os tipos de fibrilas amiloides, conforme exposto na Tabela 1.
A amiloidose primária
sistêmica (tipo AL) é uma discrasia de célula plasmática com
envolvimento multissistêmico, progressão rápida e sobrevida curta. As
fibrilas de amiloide são compostas de cadeia leve monoclonal. A mais
encontrada é a lambda, numa proporção de 2:1 em relação à kappa.
Usualmente afeta indivíduos com mais de 40 anos de idade, é mais
frequente em homens (65%) e os sintomas e sinais são frequentemente
inespecíficos, tais como fraqueza, fadiga e perda ponderal.
As
principais repercussões clínicas, que alertam para o diagnóstico,
comprometem rins, pulmões e coração e são reconhecidas tardiamente, por
isso apresentam pior evolução. A gravidade está relacionada ao aumento
dos órgãos secundários ao depósito de amiloide. Nos rins, a proteinúria,
por vezes nefrótica, é encontrada em 1/3 dos pacientes e frequentemente
leva à insuficiência renal. Em aproximadamente 1/4 dos casos
identifica-se insuficiência cardíaca congestiva, miocardiopatia
restritiva, baixa voltagem ao eletrocardiograma e hipersensibilidade
digitálica, responsáveis por 50% dos óbitos nesta forma de amiloidose. O
depósito pulmonar ocorre em aproximadamente 90% dos casos e determina
quadro de dispneia progressiva. O comprometimento hepático normalmente
não leva à perda de função, apesar de levar à hepatomegalia. Alterações
gastrintestinais como má absorção, diarreia e obstrução também podem ser
encontradas.
Um
sinal fácil de observação e que apresenta um importante valor preditivo
na doença é a presença de macroglossia, observada em até 20% dos casos,
e também frente a sangramentos devido à deficiência de fator X. Além
disso, o acometimento cutâneo caracterizado por púrpura, pápulas ou
tumores pode ser encontrado em mais de 40% dos casos. Aproximadamente 10 a 15% dos pacientes podem apresentar ainda neuropatia periférica e hipotensão ortostática.
Os
achados reumatológicos mais significantes são o aparecimento de
síndrome do túnel do carpo, observado em mais de 20% dos casos, e o
sinal de shoulder pad, que corresponde ao depósito de amiloide nos
ombros. A artropatia não é frequente, ocorre em menos de 5% dos casos e
predomina nas grandes articulações.
A
presença de um dos sintomas descritos associado à proteína monoclonal
sérica ou urinária constitui forte suspeita de amiloidose; a presença de
proteína monoclonal no soro ou urina é observada em 90% dos casos.
A amiloidose associada ao mieloma múltiplo
(tipo AL) também apresenta fibrilas compostas de cadeia leve
monoclonal, assim como a amiloidose primária. Porém, nesta forma, existe
um predomínio da cadeia leve kappa em relação à cadeia lambda, numa
relação de 2:1. As lesões osteolíticas encontradas no mieloma são de
grande auxílio no seu diferencial, visto que as manifestações clínicas
são semelhantes àquelas encontradas na forma primária.
A amiloidose secundária ou reativa ou adquirida
(tipo AA) é uma complicação pouco comum de estados inflamatórios
crônicos, infecções e, de forma mais rara, a determinados tipos de
neoplasias. A forma reativa a processos inflamatórios crônicos tem sido
relacionada principalmente às doenças reumatológicas, particularmente a
artrite reumatoide (do adulto e juvenil), espondilite anquilosante,
síndrome de Reiter e artrite psoriática. Interessante notar que, apesar
disso, a amiloidose é rara nas doenças do tecido conectivo,
principalmente no lúpus eritematoso sistêmico. Na doença de Chron, a
amiloidose ocorre em até 8% dos casos, em contraste com a retocolite
ulcerativa, onde é extremamente rara.
A
suspeita de amiloidose nestas patologias fundamenta-se basicamente na
presença de proteinúria, má absorção intestinal ou envolvimento
gastrintestinal como náusea, diarreia e hepato ou esplenomegalia. O
depósito amiloide é o da proteína AA que é distinto das cadeias leves de
imunoglobulinas, e os pacientes raramente têm acometimento cardíaco e
de nervo periférico. A macroglossia não é uma característica da
amiloidose AA. O tratamento desta forma secundária de amiloidose
consiste no controle do processo inflamatório da doença de base, levando
a estabilização ou resolução parcial do depósito amiloide.
A amiloidose heredofamiliar
é identificada pelas várias regiões onde cada família e tipos de
depósito amiloide foram descritos e estão associados a quadros clínicos
distintos. O modo de transmissão habitualmente é autossômico dominante,
com a exceção da febre familiar do Mediterrâneo, que é autossômica
recessiva. Esta se caracteriza por surtos recorrentes de febre,
serosites (principalmente peritonite e pleurite) e artrite, que se
inicia na infância/adolescência, principalmente nos de origem
mediterrânea. O depósito amiloide é do tipo AA, semelhante ao das formas
reativas, e envolve fígado, baço, suprarrenal e principalmente rins,
onde a proteinúria é inicialmente observada e, após 2 a 15 anos, evolui invariavelmente para síndrome nefrótica e insuficiência renal.
Outras Formas de Amiloidose
A amiloidose local
pode estar presente em qualquer órgão, por vezes assemelhando-se a
tumores, principalmente em pulmão, pele, laringe, olhos e vesícula.
A amiloidose associada à hemodiálise crônica
é um grande desafio no tratamento da insuficiência renal crônica, pois,
nesta forma, existe o depósito de fibrilas constituídas por b2
microglobulina intacta que normalmente não é removida durante a
diálise. A sua incidência é superior a 50% após 5 anos do início desse
procedimento. Caracteriza-se por quadro de poliartrite tipo reumatoide
com formação de pannus, tenossinovite, síndrome do túnel do carpo e
aparecimento de grandes cistos e erosões ósseas, eventualmente levando a
fraturas patológicas. Até o momento, o transplante renal é o único
tratamento efetivo desta condição, pois leva à rápida normalização dos
níveis séricos de beta-2-microglobulina e resolução dos sintomas.
A amiloidose cerebral
está estreitamente relacionada à doença de Alzheimer, síndrome de Down e
amiloidose hereditária com hemorragia cerebral (holandesa), que
compartilham o depósito da proteína amiloide beta (A-beta). Importante
ressaltar que o cérebro e os vasos sanguíneos cerebrais são raramente
afetados nas amiloidoses sistêmicas, porém são importantes sítios de
depósito local nestas formas.
A amiloidose endócrina ocorre
em glândulas secretoras de hormônios polipeptídicos, bem como em
tumores secretores hormonais. No diabetes mellitus não
insulino-dependente, o depósito de proteína amiloide do tipo AIAPP
(polipeptídio amiloide das ilhotas de Langerhans) pode ser encontrado em
até 95% dos pacientes com esta doença.
Tabela 1: Formas clínicas de amiloidose
|
amiloidose
|
Proteína amiloide
|
Condições associadas
|
|
Primária
|
AL
|
Sem doença prévia ou concomitante
|
|
Do mieloma múltiplo
|
AL
|
Mieloma múltiplo
|
|
Secundária ou reativa
|
AA
|
Infecções crônicas: mal de Hansen, tuberculose, osteomielite
|
|
AA
|
Inflamações crônicas: doença reumatoide do adulto, doença reumatoide juvenil, espondilite anquilosante
| |
|
Heredofamiliar
|
AA
|
Febre familiar do Mediterrâneo
|
|
AA
|
Síndrome de Muckle-Well
| |
|
ATTR
|
Neuropática
| |
|
ATTR
|
Cardíaca
| |
|
AGel
|
Distrofia corneana familiar amiloide
| |
|
Localizada
|
AL
|
Sem comprometimento sistêmico
|
|
Da hemodiálise
|
Ab2M
|
Hemodiálise crônica
|
|
Cerebral
|
Ab
|
Doença de Alzheimer
|
|
Ab
|
Síndrome de Down
| |
|
ACys
|
Hereditária com hemorragia cerebral
| |
|
AScr
|
Doença de Creutzfeldt-Jakob
| |
|
Endócrina
|
ACal
|
Carcinoma medular de tireoide
|
|
AIAPP
|
Diabetes mellitus
|
MANIFESTAÇÕES REUMATOLÓGICAS DA AMILOIDOSE
A
substância amiloide é depositada na articulação, na membrana e no
líquido sinovial. A artropatia amiloide é encontrada principalmente na
amiloidose secundária ao mieloma múltiplo, mas também na forma primária,
na febre familiar do Mediterrâneo e na forma associada à diálise.
A
artrite amiloide geralmente mimetiza aquela encontrada na artrite
reumatoide, com quadro simétrico, de pequenas articulações, associado a
rigidez matinal e fadiga. As articulações preferencialmente
comprometidas são ombros, punhos, joelhos e dedos; elas se apresentam
edemaciadas e firmes, mas sem rubor e dor importante. Neste sentido, um
importante espessamento sinovial deve ser considerado como indicativo de
amiloidose.
A
artropatia amiloide pode manifestar-se como síndrome do tunel do carpo
devido à infiltração de punhos, principalmente em indivíduos com mais de
50 anos de idade. Na amiloidose primária, a síndrome do túnel do carpo
pode ser encontrada em até 20% dos casos e pode ser decisiva para o seu
diagnóstico. Contudo, esta é mais frequente na amiloidose secundária à
hemodiálise crônica, onde a compressão do nervo mediano ocorre em mais
de 70% dos pacientes. Outra articulação que merece destaque é o ombro. O
shoulder pad ocorre principalmente na amiloidose primária em
decorrência da intensa infiltração amiloide em ombros. Também é
importante sinal sugestivo de diagnóstico da amiloidose.
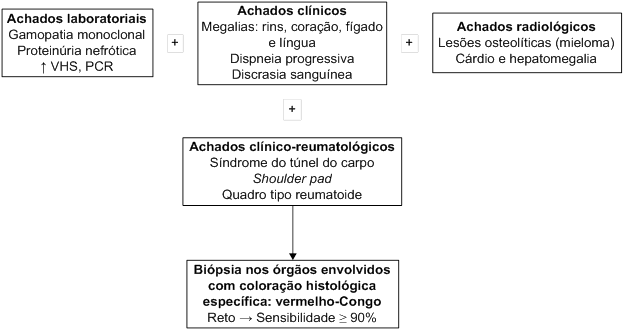
DIAGNÓSTICO E EXAMES COMPLEMENTARES
Na
maioria das vezes, a suspeita clínica é secundária ao depósito amiloide
nos órgãos afetados. Portanto, a investigação deve ser dirigida
mediante o sítio comprometido. As alterações nos exames de sangue e
urina são, por vezes, inespecíficos, como um aumento de velocidade de
hemossedimentação (VHS) e da PCR. A presença de proteinúria e
hipergamaglobulinemia pode levar a suspeita de amiloidose AL,
principalmente quando associadas a neuropatia periférica ou
insuficiência cardíaca congestiva. Nas formas de amiloidose primária
sistêmica e naquela associada ao mieloma múltiplo, a identificação de
uma cadeia leve monoclonal pode auxiliar na suspeita diagnóstica, embora
possa ainda ser negativa em 15 a
20% dos casos. A imunoeletroforese revela a presença de cadeia leve
monoclonal, frequentemente lambda, na amiloidose primária sistêmica
(tipo AL) ou da cadeia leve kappa na amiloidose secundária ao mieloma
múltiplo.
O
diagnóstico de amiloidose baseia-se na demonstração histológica do
depósito tecidual de amiloide por meio de coloração específica. A
confirmação histológica do amiloide é classicamente demonstrada pela
coloração com vermelho-Congo, que, sob microscopia polarizada, apresenta
uma birrefringência verde. Nem sempre as fibrilas amiloides são
identificadas à microscopia eletrônica e, na verdade, seu achado isolado
também não permite a confirmação diagnóstica.
A
biópsia deve ser preferencialmente realizada no órgão suspeito de
infiltração amiloide, considerando-se que existe o risco de complicações
relacionadas a sangramento. A biópsia de reto ou de gordura subcutânea
são as menos invasivas, com positividade de mais de 90% tanto nas formas
primárias quanto secundárias. Outros sítios passíveis de biópsias são a
gengiva e a medula óssea, que possuem sensibilidades de 80% e 50%,
respectivamente. O uso de anticorpos dirigidos contra as proteínas
fibrilares amiloides, por imunofluorescência e por imunoperoxidase, pode
facilitar a sua identificação. Porém, a positividade à
imuno-histoquímica do amiloide não constitui método isolado de
diagnóstico, havendo também a necessidade da confirmação pela coloração
com vermelho-Congo.
Radiologicamente,
a osteopenia generalizada é observada em até 80% dos casos, mesmo na
ausência de lesões líticas. O fator reumatoide habitualmente é negativo,
sendo de auxílio na sua diferenciação. A análise do líquido sinovial
revela um líquido não inflamatório com predomínio de mononucleares e
presença de depósitos amiloides no vilo sinovial à coloração do
vermelho-Congo.
TRATAMENTO
Os
princípios da terapêutica da amiloidose baseiam-se na redução da
formação do amiloide e, ao mesmo tempo, dissolução dos depósitos
existentes.
A
redução do precursor amiloide consiste na diminuição da síntese
hepática das proteínas de fase aguda. De fato, tem sido demonstrado que a
utilização de drogas citotóxicas na artrite reumatoide juvenil e do
adulto reduziu a incidência de amiloidose e melhorou o prognóstico
dessas doenças.
O
clássico uso da colchicina no tratamento da febre familiar do
Mediterrâneo, além de prevenir ataques febris, também se mostrou
comprovadamente efetivo na redução do depósito amiloide, preservando
função renal e aumentando a sobrevida ao longo do tempo. A colchicina
também parece exercer efeitos adicionais na amiloidose AL,
principalmente quando associada a melfalam e prednisolona. A realização
de plasmaférese e o transplante hepático também parecem ser efetivos
como redutores da amiloidose familiar por TTR, assim como o transplante
renal para a amiloidose causada pela beta-2-microglobulina (A-beta-2M).
Medidas
de suporte são particularmente importantes no seguimento destes
pacientes, por retardar a piora da insuficiência de um determinado
órgão, por exemplo, o controle rigoroso dos níveis pressóricos na
amiloidose renal. O transplante hepático, renal e cardíaco pode ser
necessário em casos extremos, sobretudo na amiloidose do tipo AA. A
utilização de determinadas drogas, como digital e bloqueadores de
cálcio, deve ser cuidadosa nos casos com envolvimento cardíaco, pois
podem agravar a função miocárdica.
Até
o momento, a história natural da amiloidose sistêmica apresenta
prognóstico e desfecho ruins, principalmente devido ao início tardio do
tratamento. O reconhecimento precoce dessa doença, prevenindo o depósito
de amiloide nos órgãos, é um dos fatores mais importantes para sua boa
evolução
ALGORITMO
Algoritmo 1: Fluxograma para amiloidose

BIBLIOGRAFIA
1. Benson MD. Familial amyloidosis. Journal of Internal Medicine 1992; 232:525-7.
2. Cohen
AS. Amyloidosis. In: McCarty DJ, Koopman WJ. Arthritis and allied
conditions. 12. ed. Philadelphia: Lea & Febiger, 1993. p.1427-47.
3. Gertz MA, Kyle RA. Amyloidosis: prognosis and treatment. Seminars in Arthritis and Rheumatism 1994; 24:124-38.
4. Gertz MA. Secondary amyloidosis (AA). Journal of Internal Medicine 1992; 232:517-8.
5. Husby G. Amyloidosis. Seminars in Arthritis and Rheumatism 1992; 22:67-82.
6. Husby G. Nomenclature and classification of amyloid and amyloidoses. Journal of Internal Medicine 1992; 232:511-2.
7. Kisilevsky
R. Proteoglycans, glycosaminoglycans, amyloid-enhancing factor, and
amyloid deposition. Journal of Internal Medicine 1992; 232:515-6.
8. Kyle RA. Primary systemic amyloidosis. Journal of Internal Medicine 1992; 232:523-4.
9. Livneh
A, Zener D, Langevitz P, Shemer J, Sohar E, Pras M. Colchicine in the
treatment of AA and AL amyloidosis. Seminars in Arthritis and Rheumatism
1993; 23:206-14.
10. Pepys MB. Amyloid P component and the diagnosis of amyloidosis. Journal of Internal Medicine 1992; 232:519-21.
11. Skinner M. Protein AA/SAA. Journal of Internal Medicine 1992; 232:513-4.
12. Tan SY, Pepys MB. Amyloidosis. Histopathology 1994; 25:403-14.
Sem comentários:
Enviar um comentário